Auxiliary files
Auxiliary readers allow you to read in timeseries data accompanying a trajectory that is not stored in the regular trajectory file.
Supported formats
Reader
Format
Extension (if file)
Remarks
XVGReaderXVG
xvg
(default)
Produced by Gromacs during simulation or analysis. Reads full file on initialisation.
XVGFileReaderXVG-F
xvg
Alternate xvg file reader, reading each step from the file in turn for a lower memory footprint.
XVGReaderis the default reader for .xvg files.
EDRReaderEDR
edr
Produced by Gromacs during simulation or analysis. Reads full file on initialisation.
XVG Files
Reading data directly
In [1]: import MDAnalysis as mda
In [2]: from MDAnalysis.tests.datafiles import XVG_BZ2 # cobrotoxin protein forces
In [3]: aux = mda.auxiliary.core.auxreader(XVG_BZ2)
In [4]: aux
Out[4]: <MDAnalysis.auxiliary.XVG.XVGReader at 0x7f96f0621250>
In stand-alone use, an auxiliary reader allows you to iterate over each step in a set of auxiliary data.
In [5]: for step in aux:
...: print(step.data)
...:
[ 0. 200.71288 -1552.2849 ... 128.4072 1386.0378
-2699.3118 ]
[ 50. -1082.6454 -658.32166 ... -493.02954 589.8844
-739.2124 ]
[ 100. -246.27269 146.52911 ... 484.32501 2332.3767
-1801.6234 ]
Use slicing to skip steps.
In [6]: for step in aux[1:2]:
...: print(step.time)
...:
50.0
The auxreader() function uses the get_auxreader_for() to return an appropriate class. This can guess the format either from a filename, ‘
In [7]: mda.auxiliary.core.get_auxreader_for(XVG_BZ2)
Out[7]: MDAnalysis.auxiliary.XVG.XVGReader
or return the reader for a specified format.
In [8]: mda.auxiliary.core.get_auxreader_for(format='XVG-F')
Out[8]: MDAnalysis.auxiliary.XVG.XVGFileReader
Loading data into a Universe
Auxiliary data may be added to a trajectory Reader through the
add_auxiliary() method. Auxiliary data
may be passed in as a AuxReader instance, or directly as e.g. a filename, in
which case get_auxreader_for() is used to
guess an appropriate reader.
In [9]: from MDAnalysis.tests.datafiles import PDB_xvf, TRR_xvf
In [10]: u = mda.Universe(PDB_xvf, TRR_xvf)
In [11]: u.trajectory.add_auxiliary('protein_force', XVG_BZ2)
In [12]: for ts in u.trajectory:
....: print(ts.aux.protein_force)
....:
[ 0. 200.71288 -1552.2849 ... 128.4072 1386.0378
-2699.3118 ]
[ 50. -1082.6454 -658.32166 ... -493.02954 589.8844
-739.2124 ]
[ 100. -246.27269 146.52911 ... 484.32501 2332.3767
-1801.6234 ]
Passing arguments to auxiliary data
For alignment with trajectory data, auxiliary readers provide methods to assign each auxiliary step to the nearest trajectory timestep, read all steps assigned to a trajectory timestep and calculate ‘representative’ value(s) of the auxiliary data for that timestep.
To set a timestep or ??
‘Assignment’ of auxiliary steps to trajectory timesteps is determined from the time
of the auxiliary step, dt of the trajectory and time at the first frame of the
trajectory. If there are no auxiliary steps assigned to a given timestep (or none within
cutoff, if set), the representative value(s) are set to np.nan.
Iterating over auxiliary data
Auxiliary data may not perfectly line up with the trajectory, or have missing data.
In [13]: from MDAnalysis.tests.datafiles import PDB, TRR
In [14]: u_long = mda.Universe(PDB, TRR)
In [15]: u_long.trajectory.add_auxiliary('protein_force', XVG_BZ2, dt=200)
In [16]: for ts in u_long.trajectory:
....: print(ts.time, ts.aux.protein_force[:4])
....:
0.0 [ 0. 200.71288 -1552.2849 -967.21124]
100.00000762939453 [ 100. -246.27269 146.52911 -1084.2484 ]
200.00001525878906 [nan nan nan nan]
300.0 [nan nan nan nan]
400.0000305175781 [nan nan nan nan]
500.0000305175781 [nan nan nan nan]
600.0 [nan nan nan nan]
700.0000610351562 [nan nan nan nan]
800.0000610351562 [nan nan nan nan]
900.0000610351562 [nan nan nan nan]
The trajectory ProtoReader methods
next_as_aux() and
iter_as_aux() allow for movement
through only trajectory timesteps for which auxiliary data is available.
In [17]: for ts in u_long.trajectory.iter_as_aux('protein_force'):
....: print(ts.time, ts.aux.protein_force[:4])
....:
0.0 [ 0. 200.71288 -1552.2849 -967.21124]
100.00000762939453 [ 100. -246.27269 146.52911 -1084.2484 ]
This may be used to
avoid representative values set to np.nan, particularly when auxiliary data
is less frequent.
Sometimes the auxiliary data is longer than the trajectory.
In [18]: u_short = mda.Universe(PDB)
In [19]: u_short.trajectory.add_auxiliary('protein_force', XVG_BZ2)
In [20]: for ts in u_short.trajectory:
....: print(ts.time, ts.aux.protein_force)
....:
0.0 [ 0. 200.71288 -1552.2849 ... 128.4072 1386.0378
-2699.3118 ]
In order to acess auxiliary values at every individual step, including those
outside the time range of the trajectory,
iter_auxiliary() allows iteration
over the auxiliary independent of the trajectory.
In [21]: for step in u_short.trajectory.iter_auxiliary('protein_force'):
....: print(step.data)
....:
[ 0. 200.71288 -1552.2849 ... 128.4072 1386.0378
-2699.3118 ]
[ 50. -1082.6454 -658.32166 ... -493.02954 589.8844
-739.2124 ]
[ 100. -246.27269 146.52911 ... 484.32501 2332.3767
-1801.6234 ]
To iterate over only a certain section of the auxiliary:
In [22]: for step in u_short.trajectory.iter_auxiliary('protein_force', start=1, step=2):
....: print(step.time)
....:
50.0
The trajectory remains unchanged, and the auxiliary will be returned to the current timestep after iteration is complete.
Accessing auxiliary attributes
To check the values of attributes of an added auxiliary, use
get_aux_attribute().
In [23]: u.trajectory.get_aux_attribute('protein_force', 'dt')
Out[23]: 50.0
If attributes are settable, they can be changed using
set_aux_attribute().
In [24]: u.trajectory.set_aux_attribute('protein_force', 'data_selector', [1])
The auxiliary may be renamed using set_aux_attribute with ‘auxname’, or more
directly by using rename_aux().
In [25]: u.trajectory.ts.aux.protein_force
Out[25]:
array([ 0. , 200.71288, -1552.2849 , ..., 128.4072 ,
1386.0378 , -2699.3118 ])
In [26]: u.trajectory.rename_aux('protein_force', 'f')
In [27]: u.trajectory.ts.aux.f
Out[27]:
array([ 0. , 200.71288, -1552.2849 , ..., 128.4072 ,
1386.0378 , -2699.3118 ])
Recreating auxiliaries
To recreate an auxiliary, the set of attributes necessary to replicate it can
first be obtained with get_description().
The returned dictionary can then be passed to
auxreader() to load a new copy of the
original auxiliary reader.
In [28]: description = aux.get_description()
In [29]: list(description.keys())
Out[29]:
['represent_ts_as',
'cutoff',
'dt',
'initial_time',
'time_selector',
'data_selector',
'constant_dt',
'auxname',
'format',
'auxdata']
In [30]: del aux
In [31]: mda.auxiliary.core.auxreader(**description)
Out[31]: <MDAnalysis.auxiliary.XVG.XVGReader at 0x7f96f0ac9640>
The ‘description’ of any or all the auxiliaries added to a trajectory can be
obtained using get_aux_descriptions().
In [32]: descriptions = u.trajectory.get_aux_descriptions(['f'])
To reload, pass the dictionary into add_auxiliary().
In [33]: u2 = mda.Universe(PDB, TRR)
In [34]: for desc in descriptions:
....: u2.trajectory.add_auxiliary(**desc)
....:
EDR Files
EDR files are created by GROMACS during simulations and contain additional non-trajectory time-series data of the system, such as energies, temperature, or pressure. The EDRReader allows direct reading of these binary files and associating of the data with trajectory time steps just like the XVGReader does. As all functionality of the base AuxReaders (see XVGReader above) also works with the EDRReader, this section will highlight functionality unique to the EDRReader.
Standalone Usage
The EDRReader is initialised by passing the path to an EDR file as an argument.
In [35]: import MDAnalysis as mda
In [36]: from MDAnalysisTests.datafiles import AUX_EDR
In [37]: aux = mda.auxiliary.EDR.EDRReader(AUX_EDR)
# Or, for example
# aux = mda.auxiliary.EDR.EDRReader("path/to/file/ener.edr")
Dozens of terms can be defined in EDR files. A list of available data is conveniently available unter the terms attribute.
In [38]: aux.terms
Out[38]:
['Time',
'Bond',
'Angle',
'Proper Dih.',
'Ryckaert-Bell.',
'LJ-14',
'Coulomb-14',
'LJ (SR)',
'Disper. corr.',
'Coulomb (SR)',
'Coul. recip.',
'Potential',
'Kinetic En.',
'Total Energy',
'Conserved En.',
'Temperature',
'Pres. DC',
'Pressure',
'Constr. rmsd',
'Box-X',
'Box-Y',
'Box-Z',
'Volume',
'Density',
'pV',
'Enthalpy',
'Vir-XX',
'Vir-XY',
'Vir-XZ',
'Vir-YX',
'Vir-YY',
'Vir-YZ',
'Vir-ZX',
'Vir-ZY',
'Vir-ZZ',
'Pres-XX',
'Pres-XY',
'Pres-XZ',
'Pres-YX',
'Pres-YY',
'Pres-YZ',
'Pres-ZX',
'Pres-ZY',
'Pres-ZZ',
'#Surf*SurfTen',
'Box-Vel-XX',
'Box-Vel-YY',
'Box-Vel-ZZ',
'T-Protein',
'T-non-Protein',
'Lamb-Protein',
'Lamb-non-Protein']
To extract data for plotting, the get_data method can be used. It can be used to extract a single, multiple, or all terms as follows:
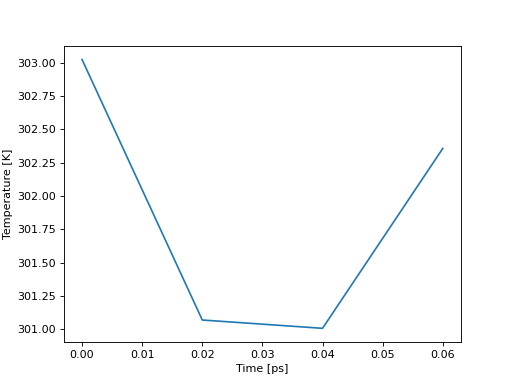
In [39]: temp = aux.get_data("Temperature")
In [40]: print(temp.keys())
dict_keys(['Time', 'Temperature'])
In [41]: energies = aux.get_data(["Potential", "Kinetic En."])
In [42]: print(energies.keys())
dict_keys(['Time', 'Potential', 'Kinetic En.'])
In [43]: all_data = aux.get_data()
In [44]: print(all_data.keys())
dict_keys(['Time', 'Bond', 'Angle', 'Proper Dih.', 'Ryckaert-Bell.', 'LJ-14', 'Coulomb-14', 'LJ (SR)', 'Disper. corr.', 'Coulomb (SR)', 'Coul. recip.', 'Potential', 'Kinetic En.', 'Total Energy', 'Conserved En.', 'Temperature', 'Pres. DC', 'Pressure', 'Constr. rmsd', 'Box-X', 'Box-Y', 'Box-Z', 'Volume', 'Density', 'pV', 'Enthalpy', 'Vir-XX', 'Vir-XY', 'Vir-XZ', 'Vir-YX', 'Vir-YY', 'Vir-YZ', 'Vir-ZX', 'Vir-ZY', 'Vir-ZZ', 'Pres-XX', 'Pres-XY', 'Pres-XZ', 'Pres-YX', 'Pres-YY', 'Pres-YZ', 'Pres-ZX', 'Pres-ZY', 'Pres-ZZ', '#Surf*SurfTen', 'Box-Vel-XX', 'Box-Vel-YY', 'Box-Vel-ZZ', 'T-Protein', 'T-non-Protein', 'Lamb-Protein', 'Lamb-non-Protein'])
The times of each data point are always part of the returned dictionary to facilicate plotting.
In [45]: import matplotlib.pyplot as plt
In [46]: plt.plot(temp["Time"], temp["Temperature"])
Out[46]: [<matplotlib.lines.Line2D at 0x7f96e36c6190>]
In [47]: plt.ylabel("Temperature [K]")
Out[47]: Text(0, 0.5, 'Temperature [K]')
In [48]: plt.xlabel("Time [ps]")
Out[48]: Text(0.5, 0, 'Time [ps]')
In [49]: plt.show()
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
Unit Handling
On object creation, the EDRReader creates a unit_dict attribute which contains
information on the units of the data stored within. These units are read from the
EDR file automatically and by default converted to MDAnalysis base units where such units
are defined. The automatic unit conversion can be disabled by setting the convert_units kwarg to False.
In [50]: aux.unit_dict["Box-X"]
Out[50]: 'A'
In [51]: aux_native = mda.auxiliary.EDR.EDRReader(AUX_EDR, convert_units=False)
In [52]: aux_native.unit_dict["Box-X"]
Out[52]: 'nm'
Use with Trajectories
An arbitrary number of terms to be associated with a trajectory can be specified as a dictionary. The dictionary is chosen so that the name to be used in MDAnalysis to access the data is mapped to the name in aux.terms.
In [53]: from MDAnalysisTests.datafiles import AUX_EDR_TPR, AUX_EDR_XTC
In [54]: u = mda.Universe(AUX_EDR_TPR, AUX_EDR_XTC)
In [55]: term_dict = {"temp": "Temperature", "epot": "Potential"}
In [56]: u.trajectory.add_auxiliary(term_dict, aux)
In [57]: u.trajectory.ts.aux.epot
Out[57]: -525164.0625
Adding all data is possible by omitting the dictionary as follows. It is then necessary to specify that the EDRReader should be passed as the auxdata argument as such:
In [58]: u = mda.Universe(AUX_EDR_TPR, AUX_EDR_XTC)
In [59]: u.trajectory.add_auxiliary(auxdata=aux)
In [60]: u.trajectory.aux_list
Out[60]: dict_keys(['Time', 'Bond', 'Angle', 'Proper Dih.', 'Ryckaert-Bell.', 'LJ-14', 'Coulomb-14', 'LJ (SR)', 'Disper. corr.', 'Coulomb (SR)', 'Coul. recip.', 'Potential', 'Kinetic En.', 'Total Energy', 'Conserved En.', 'Temperature', 'Pres. DC', 'Pressure', 'Constr. rmsd', 'Box-X', 'Box-Y', 'Box-Z', 'Volume', 'Density', 'pV', 'Enthalpy', 'Vir-XX', 'Vir-XY', 'Vir-XZ', 'Vir-YX', 'Vir-YY', 'Vir-YZ', 'Vir-ZX', 'Vir-ZY', 'Vir-ZZ', 'Pres-XX', 'Pres-XY', 'Pres-XZ', 'Pres-YX', 'Pres-YY', 'Pres-YZ', 'Pres-ZX', 'Pres-ZY', 'Pres-ZZ', '#Surf*SurfTen', 'Box-Vel-XX', 'Box-Vel-YY', 'Box-Vel-ZZ', 'T-Protein', 'T-non-Protein', 'Lamb-Protein', 'Lamb-non-Protein'])
Selecting Trajectory Frames Based on Auxiliary Data
One use case for the new auxiliary readers is the selection of frames based on auxiliary data. To select only those frames with a potential energy below a certain threshold, the following can be used:
In [61]: u = mda.Universe(AUX_EDR_TPR, AUX_EDR_XTC)
In [62]: term_dict = {"epot": "Potential"}
In [63]: u.trajectory.add_auxiliary(term_dict, aux)
In [64]: selected_frames = np.array([ts.frame for ts in u.trajectory if ts.aux.epot < -524600])
A slice of the trajectory can then be obtained from the list of frames as such:
In [65]: trajectory_slice = u.trajectory[selected_frames]
In [66]: print(len(u.trajectory))
4
In [67]: print(len(trajectory_slice))
2
Memory Usage
It is assumed that the EDR file is small enough to be read in full. However, since one EDRReader instance is created for each term added to a trajectory, memory usage monitoring was implemented. A warning will be issued if 1 GB of memory is used by auxiliary data. This warning limit can optionally be changed by defining the memory_limit (in bytes) when adding data to a trajectory. Below, the memory limit is set to 200 MB.
In [68]: u = mda.Universe(AUX_EDR_TPR, AUX_EDR_XTC)
In [69]: term_dict = {"temp": "Temperature", "epot": "Potential"}
In [70]: u.trajectory.add_auxiliary(term_dict, aux, memory_limit=2e+08)