Contributing to the main codebase
If you would like to contribute, start by searching through the issues and pull requests to see whether someone else has raised a similar idea or question.
If you don’t see your idea or problem listed, do one of the following:
If your contribution is minor, such as a typo fix, go ahead and fix it by following the guide below and open a pull request.
If your contribution is major, such as a bug fix or a new feature, start by opening an issue first. That way, other people can weigh in on the discussion before you do any work. If you also create a pull request, you should link it to the issue by including the issue number in the pull request’s description.
Here is an overview of the development workflow for code or inline code documentation, as expanded on throughout the rest of the page.
Fork the MDAnalysis repository from the mdanalysis account into your own account
Set up an isolated virtual environment for code development
Build development versions of MDAnalysis and MDAnalysisTests on your computer into the virtual environment
Add your new feature or bug fix or add your new documentation
Add and run tests (if adding to the code)
Build and view the documentation (if adding to the docs)
Ensure PEP8 compliance (mandatory) and format your code with Darker (optional)
Working with the code
Forking
You will need your own fork to work on the code. Go to the MDAnalysis project page and hit the Fork button. You will want to clone your fork to your machine:
git clone https://github.com/your-user-name/mdanalysis.git
cd mdanalysis
git remote add upstream https://github.com/MDAnalysis/mdanalysis
This creates the directory mdanalysis and connects your repository to the upstream (main project) MDAnalysis repository.
Creating a development environment
To change code and test changes, you’ll need to build both MDAnalysis and MDAnalysisTests from source. This requires a Python environment. We highly recommend that you use virtual environments. This allows you to have multiple experimental development versions of MDAnalysis that do not interfere with each other, or your own stable version. Since MDAnalysis is split into the actual package and a test suite, you need to install both modules in development mode.
You can do this either with conda or pip.
Note
If you are a first time contributor and/or don’t have a lot of experience managing
your own Python virtual environments, we strongly suggest using conda.
You only need to follow the sections corresponding to the installation method you
choose.
With conda-forge packages
We will use pre-compiled packages from the conda-forge repository.
The program to manage these packages is called conda although for the following we recommend using the faster one called mamba. We will use mamba for the examples but you can equally use conda instead.
For
mamba, follow the mamba installation instructions or the miniforge installation instructions.For
condainstall either the Anaconda distribution or miniconda.
Make sure your mamba (or conda) is up to date:
mamba update mamba
Create a new environment with mamba create. This will allow you to change code in
an isolated environment without touching your base Python installation, and without
touching existing environments that may have stable versions of MDAnalysis. :
mamba create --name mdanalysis-dev "python>=3.12"
Use a recent version of Python that is supported by MDAnalysis for this environment.
Activate the environment to build MDAnalysis into it:
mamba activate mdanalysis-dev
Warning
Make sure the mdanalysis-dev environment is active when developing MDAnalysis.
To view your environments:
mamba info -e
To list the packages installed in your current environment:
mamba list
Note
When you finish developing MDAnalysis you can deactivate the environment with
mamba deactivate, in order to return to your root environment.
See the full mamba documentation or the full conda documentation for more details.
With pip and virtualenv
Like conda, virtual environments managed with virtualenv allow you to use different versions of Python and Python packages for your different project. Unlike conda, virtualenv is not a general-purpose package manager. Instead, it leverages what is available on your system, and lets you install Python packages using pip.
To use virtual environments you have to install the virtualenv package first. This can be done with pip:
python -m pip install virtualenv
Virtual environments can be created for each project directory.
cd my-project/ virtualenv my-project-env
This will create a new folder my-project-env. This folder contains the virtual environment and all packages you have installed in it. To activate it in the current terminal run:
source myproject-env/bin/activate
Now you can install packages via pip without affecting your global environment. The packages that you install when the environment is activated will be available in terminal sessions that have the environment activated.
Note
When you finish developing MDAnalysis you can deactivate the environment with
deactivate, in order to return to your root environment.
The virtualenvwrapper package makes virtual environments easier to use. It provides some very useful features:
it organises the virtual environment into a single user-defined directory, so they are not scattered throughout the file system;
it defines commands for the easy creation, deletion, and copying of virtual environments;
it defines a command to activate a virtual environment using its name;
all commands defined by
virtualenvwrapperhave tab-completion for virtual environment names.
You first need to install virtualenvwrapper outside of a virtual environment:
python -m pip install virtualenvwrapper
Then, you need to load it into your terminal session. Add the following lines in ~/.bashrc. They will be executed every time you open a new terminal session:
# Decide where to store the virtual environments export WORKON_HOME=~/Envs # Make sure the directory exists mkdir -p ${WORKON_HOME} # Load virtualenvwrapper source /usr/local/bin/virtualenvwrapper.sh
Open a new terminal or run source ~/.bashrc to update your session. You can now create a virtual environment with:
mkvirtualenv my-project
Regardless of your current working directory, the environment is created in ~/Envs/ and it is now loaded in our terminal session.
You can load your virtual environments by running workon my-project, and exit them by running deactivate.
Virtual environments, especially with virtualenvwrapper, can do much more. For example, you can create virtual environments with different python interpreters with the -p flag. The Hitchhiker’s Guide to Python has a good tutorial that gives a more in-depth explanation of virtual environments. The virtualenvwrapper documentation is also a good resource to read.
On a Mac
One more step is often required on macOS, because of the default number of files that a process can open simultaneously is quite low (256). To increase the number of files that can be accessed, run the following command:
ulimit -n 4096
This sets the number of files to 4096. However, this command only applies to your currently open terminal session. To keep this high limit, add the above line to your ~/.profile.
Building MDAnalysis
With mamba/conda
Note
Make sure that you have cloned the repository
and activated your virtual environment with mamba activate mdanalysis-dev.
First we need to install the dependencies. To install the base MDAnalysis dependencies, do the following:
mamba install -c conda-forge \ 'cython>=0.28' \ 'filelock' \ 'griddataformats>=0.4.0' \ 'hypothesis' \ 'matplotlib-base>=1.5.1' \ 'mdahole2-base' \ 'mda-xdrlib' \ 'mmtf-python>=1.0.0' \ 'numpy>=1.23.2' \ 'packaging' \ 'pathsimanalysis' \ 'pytest' \ 'scipy>=1.5.0' \ 'threadpoolctl' \ 'tqdm>=4.43.0' \ 'waterdynamics'
You can also install the following optional dependencies, although please note that they many not all be available for your machine type. Specifically, hole2, and distopia are only available on Linux + x86_64 machines, and openmm is not available for Windows. Simply remove any optional package that is not available for your operating system/architecture from your list.
mamba install -c conda-forge \ 'biopython>=1.80' \ 'chemfiles>=0.10' \ 'clustalw=2.1' \ 'dask>=2023.1.1' \ 'distopia>=0.2.0' \ 'duecredit' \ 'gsd>3.0.0' \ 'h5py>=2.1.0' \ 'hole2' \ 'joblib>=0.12' \ 'netcdf4' \ 'networkx' \ 'openmm' \ 'parmed' \ 'pyedr>0.7.0' \ 'pytest-xdist' \ 'pytest-cov' \ 'pytest-timeout' \ 'pytng>=0.2.3' \ 'rdkit>=2020.03.1' \ 'scikit-learn' \ 'seaborn>=0.7.0' \ 'tidynamics>1.0.0' \ 'black=24' # documentation dependencies mamba install -c conda-forge \ 'mdanalysis-sphinx-theme>=1.3.0' \ docutils \ sphinxcontrib-bibtex \ sphinx-sitemap
Ensure that you have a working C/C++ compiler (e.g. gcc or clang). You will also need Python ≥ 3.10 (which you already installed in your virtual environment). We will now install MDAnalysis.
# go to the mdanalysis source directory cd mdanalysis/ # Build and install the MDAnalysis package cd package/ python -m pip install -e . # Build and install the test suite cd ../testsuite/ python -m pip install -e .
At this point you should be able to import MDAnalysis from your locally built version. If you are running the development version, this is visible from the version number ending in -dev0. For example:
$ python # start an interpreter >>> import MDAnalysis as mda >>> mda.__version__ '2.9.0-dev0'
With pip and virtualenv
Note
Make sure that you have cloned the repository
and activated your virtual environment with source myproject-env/bin/activate
(or workon my-project if you used the virtualenvwrapper package)
First we need to install the dependencies. To install the base MDAnalysis dependencies, do the following:
python -m pip install \ 'cython>=0.28' \ 'filelock' \ 'griddataformats>=0.4.0' \ 'hypothesis' \ 'matplotlib>=1.5.1' \ 'mdahole2' \ 'mda-xdrlib' \ 'mmtf-python>=1.0.0' \ 'numpy>=1.23.2' \ 'packaging' \ 'pathsimanalysis' \ 'pytest' \ 'scipy>=1.5.0' \ 'threadpoolctl' \ 'tqdm>=4.43.0' \ 'waterdynamics' \ 'black=24'
You can also install the following optional dependencies (note that you will not be able to install all the optional dependencies as many not available via pip, e.g. clustalw):
python -m pip install \ 'biopython>=1.80' \ 'chemfiles>=0.10' \ 'dask>=2023.1.1' \ 'distopia>=0.2.0' \ 'duecredit' \ 'gsd>3.0.0' \ 'h5py>=2.1.0' \ 'joblib>=0.12' \ 'netcdf4' \ 'networkx' \ 'parmed' \ 'pyedr>0.7.0' \ 'pytest-xdist' \ 'pytest-cov' \ 'pytest-timeout' \ 'pytng>=0.2.3' \ 'rdkit>=2020.03.1' \ 'scikit-learn' \ 'seaborn>=0.7.0' \ 'tidynamics>1.0.0' # documentation dependencies python -m pip install \ 'mdanalysis-sphinx-theme>=1.3.0' \ docutils \ sphinxcontrib-bibtex \ sphinx-sitemap
Ensure that you have a working C/C++ compiler (e.g. gcc or clang). You will also need Python ≥ 3.10. We will now install MDAnalysis.
# go to the mdanalysis source directory cd mdanalysis/ # Build and install the MDAnalysis package cd package/ python -m pip install -e . # Build and install the test suite cd ../testsuite/ python -m pip install -e .
At this point you should be able to import MDAnalysis from your locally built version. If you are running the development version, this is visible from the version number ending in “-dev0”. For example:
$ python # start an interpreter >>> import MDAnalysis as mda >>> mda.__version__ '2.9.0-dev0'
Branches in MDAnalysis
The most important branch of MDAnalysis is the develop branch, to which all development code for the next release is pushed.
The develop branch can be considered an “integration” branch for including your code into the next release. Only working, tested code should be committed to this branch. All code contributions (“features”) should branch off develop. At each release, a snapshot of the develop branch is taken, packaged and uploaded to PyPi and conda-forge.
Creating a branch
The develop branch should only contain approved, tested code, so create a
feature branch for making your changes. For example, to create a branch called
shiny-new-feature from develop:
git checkout -b shiny-new-feature develop
This changes your working directory to the shiny-new-feature branch. Keep any
changes in this branch specific to one bug or feature so it is clear
what the branch brings to MDAnalysis. You can have many branches with different names
and switch in between them using the git checkout my-branch-name command.
There are several special branch names that you should not use for your feature branches:
master
develop
package-*
gh-pages
package branches are used to prepare a new production release and should be handled by the release manager only.
master is the old stable code branch and is kept protected for historical reasons.
gh-pages is where built documentation to be uploaded to github pages is held.
Writing new code
Code formatting in Python
Since version 0.11.0, we are trying to update all the code to conform with PEP8.
- Important requirements (from PEP8):
naming:
classes: CapitalClasses (i.e. capitalized nouns without spaces)
methods and functions: underscore_methods (lower case, with underscores for spaces)
We recommend that you use a Python Integrated Development Environment (IDE) (PyCharm and others) or external tools like flake8 for code linting. For integration of external tools with emacs and vim, check out elpy (emacs) and python-mode (vim).
MDAnalysis uses black`_ for automated code formatting, which is bound by the black stability policy. The configuration for `black`_ is provided in the pyproject.toml files. To format your code with `black`_, you can run the following commands:
black package black testsuite
Modules and dependencies
MDAnalysis strives to keep dependencies small and lightweight. Code outside the MDAnalysis.analysis and MDAnalysis.visualization modules should only rely on the core dependencies, which are always installed. Analysis and visualization modules can use any any package, but the package is treated as optional.
Imports in the code should follow the General rules for importing.
See also
See Module imports in MDAnalysis for more information.
Developing in Cython
The setup.py script first looks for the .c files included in the standard MDAnalysis distribution. These are not in the GitHub repository, so setup.py will use Cython to compile extensions. .pyx source files are used instead of .c files. From there, .pyx files are converted to .c files if they are newer than the already present .c files or if the --force flag is set (i.e. python setup.py build --force). End users (or developers) should not trigger the .pyx to .c conversion, since .c files delivered with source packages are always up-to-date. However, developers who work on the .pyx files will automatically trigger the conversion since .c files will then be outdated.
Place all source files for compiled shared object files into the same directory as the final shared object file.
.pyx files and cython-generated .c files should be in the same directory as the .so files. External dependent C/C++/Fortran libraries should be in dedicated src/ and include/ folders. See the following tree as an example:
MDAnalysis |--lib | |-- _distances.so | |-- distances.pyx | |-- distances.c |-- coordinates |-- _dcdmodule.so |-- src |-- dcd.c |-- include |-- dcd.h
Testing your code
MDAnalysis takes testing seriously. All code added to MDAnalysis should have tests to ensure that it works as expected; we aim for 90% coverage. See Tests in MDAnalysis for more on writing, running, and interpreting tests.
Documenting your code
Changes to the code should be reflected in the ongoing CHANGELOG. Add an entry here to document your fix, enhancement, or change. In addition, add your name to the author list. If you are addressing an issue, make sure to include the issue number.
Ensure PEP8 compliance (mandatory) and format your code with Darker (optional)
darker is a partial formatting tool that helps to reformat new or modified code
lines so the codebase progressively adapts a code style instead of doing a full reformat,
which would be a big commitment. It was designed with the black formatter in mind, hence the name.
In MDAnalysis we only require PEP8 compliance, so if you want to make sure that your PR passes the darker bot, you’ll
need both darker and flake8:
pip install darker flake8
You’ll also need the original codebase so darker can first get a diff between the current develop branch and your code.
After making your changes to your local copy of the MDAnalysis repo, add the remote repo
(here we’re naming it upstream), and fetch the content:
git remote add upstream https://github.com/MDAnalysis/mdanalysis.git
git fetch upstream
Now you can check your modifications on the package:
darker --diff -r upstream/develop package/MDAnalysis -L flake8
and the test suite:
darker --diff -r upstream/develop testsuite/MDAnalysisTests -L flake8
Darker will first suggest changes so that the new code lines comply with black’s rules, like this:
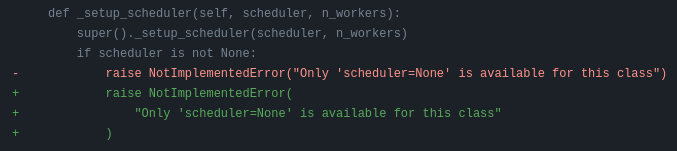
and then show flake8 errors and warnings. These look like this:
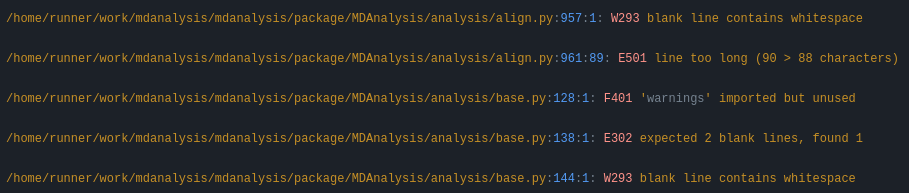
You are free to skip the diffs and then manually fix the PEP8 faults. Or if you’re ok with the suggested formatting changes, just apply the suggested fixes:
darker -r upstream/develop package/MDAnalysis -L flake8
darker -r upstream/develop testsuite/MDAnalysisTests -L flake8
Note
MDAnalysis does not currently use the popular `black`_ code formatter across the whole project, therefore please consider the following rules:
Do not run
blackon an existing file that you are editing. This makes the diff for the PR very difficult to read. Instead usedarkeras explained above to only reformat your changes.You may run
blackon a new file that you are adding to the code base.
For an ongoing discussion (which you are welcome to join) see issue #2450.
Adding your code to MDAnalysis
Committing your code
When you are happy with a set of changes and all the tests pass, it is time to commit. All changes in one revision should have a common theme. If you implemented two rather different things (say, one bug fix and one new feature), then split them into two commits with different messages.
Once you’ve made changes to files in your local repository, you can see them by typing:
git status
Tell git to track files by typing:
git add path/to/file-to-be-added.py
Doing git status again should give something like:
# On branch shiny-new-feature # # modified: /relative/path/to/file-you-added.py #
Then commit with:
git commit -m
This opens up a message editor.
Always add a descriptive comment for your commit message (feel free to be verbose!):
use a short (<50 characters) subject line that summarizes the change
leave a blank line
optionally, add additional more verbose descriptions; paragraphs or bullet lists (with
-or*) are goodmanually break lines at 80 characters
manually indent bullet lists
See also
See Tim Pope’s A Note About Git Commit Messages for a rationale for these rules.
Pushing your code to GitHub
When you want your changes to appear publicly on your GitHub page, push your forked feature branch’s commits:
git push origin shiny-new-feature
Here origin is the default name given to your remote repository on GitHub. You can see the remote repositories:
git remote -v
If you added the upstream repository as described above you will see something like:
origin [email protected]:your-username/mdanalysis.git (fetch) origin [email protected]:your-username/mdanalysis.git (push) upstream [email protected]:MDAnalysis/mdanalysis.git (fetch) upstream [email protected]:MDAnalysis/mdanalysis.git (push)
Now your code is on GitHub, but it is not yet a part of the MDAnalysis project. For that to happen, a pull request needs to be submitted on GitHub.
Rebasing your code
Often the upstream MDAnalysis develop branch will be updated while you are working on your own code. You will then need to update your own branch with the new code to avoid merge conflicts. You need to first retrieve it and then rebase your branch so that your changes apply to the new code:
git fetch upstream git rebase upstream/develop
This will replay your commits on top of the latest development code from MDAnalysis. If this
leads to merge conflicts, you must resolve these before submitting your pull
request. If you have uncommitted changes, you will need to git stash them
prior to updating. This will effectively store your changes and they can be
reapplied after updating with git stash apply.
Once rebased, push your changes:
git push -f origin shiny-new-feature
Creating a pull request
The typical approach to adding your code to MDAnalysis is to make a pull request on GitHub. Please make sure that your contribution passes all tests. If there are test failures, you will need to address them before we can review your contribution and eventually merge them. If you have problems with making the tests pass, please ask for help! (You can do this in the comments of the pull request).
Navigate to your repository on GitHub
Click on the Pull Request button
You can then click on Commits and Files Changed to make sure everything looks okay one last time
Write a description of your changes and follow the PR checklist
check that docs are updated
check that tests run
check that you’ve updated CHANGELOG
reference the issue that you address, if any
Click Send Pull Request.
Your pull request is then sent to the repository maintainers. After this, the following happens:
A suite of tests are run on your code with the tools travis, appveyor and Codecov. If they fail, please fix your pull request by pushing updates to it.
Developers will ask questions and comment in the pull request. You may be asked to make changes.
When everything looks good, a core developer will merge your code into the
developbranch of MDAnalysis. Your code will be in the next release.
If you need to make changes to your code, you can do so on your local repository as you did before. Committing and pushing the changes will update your pull request and restart the automated tests.
Working with the code documentation
MDAnalysis maintains two kinds of documentation:
This user guide: a map of how MDAnalysis works, combined with tutorial-like overviews of specific topics (such as the analyses)
The documentation generated from the code itself. Largely built from code docstrings, these are meant to provide a clear explanation of the usage of individual classes and functions. They often include technical or historical information such as in which version the function was added, or deprecation notices.
This guide is for the documentation generated from the code. If you are looking to contribute to the user guide, please see Contributing to the user guide.
MDAnalysis has a lot of documentation in the Python doc strings. The docstrings follow the Numpy Docstring Standard, which is used widely in the Scientific Python community. They are nice to read as normal text and are converted by sphinx to normal ReST through napoleon.
This standard specifies the format of the different sections of the docstring. See this document for a detailed explanation, or look at some of the existing functions to extend it in a similar manner.
Note that each page of the online documentation has a link to the Source of the page. You can look at it in order to find out how a particular page has been written in reST and copy the approach for your own documentation.
Building the documentation
The online documentation is generated from the pages in mdanalysis/package/doc/sphinx/source/documentation_pages. The documentation for the current release are hosted at www.mdanalysis.org/docs, while the development version is at www.mdanalysis.org/mdanalysis/.
In order to build the documentation, you must first clone the main MDAnalysis repo. Set up a virtual environment in the same way as you would for the code (you should typically use the same environment as you do for the code). Build the development version of MDAnalysis.
Then, generate the docs with:
cd doc/sphinx && make html
This generates and updates the files in doc/html. If the above command fails with an ImportError, run
python setup.py build_ext --inplace
and retry.
You will then be able to open the home page, doc/html/index.html, and look through the docs. In particular, have a look at any pages that you tinkered with. It is typical to go through multiple cycles of fix, rebuild the docs, check and fix again.
If rebuilding the documentation becomes tedious after a while, install the sphinx-autobuild extension.
Where to write docstrings
When writing Python code, you should always add a docstring to each public (visible to users):
module
function
class
method
When you add a new module, you should include a docstring with a short sentence describing what the module does, and/or a long document including examples and references.
Guidelines for writing docstrings
A typical function docstring looks like the following:
def func(arg1, arg2): """Summary line. Extended description of function. Parameters ---------- arg1 : int Description of `arg1` arg2 : str Description of `arg2` Returns ------- bool Description of return value """ return True
See also
The napoleon documentation has further breakdowns of docstrings at the module, function, class, method, variable, and other levels.
When writing reST markup, make sure that there are at least two blank lines above the reST after a numpy heading. Otherwise, the Sphinx/napoleon parser does not render correctly.
some more docs bla bla Notes ----- THE NEXT TWO BLANK LINES ARE IMPORTANT. .. versionadded:: 0.16.0
Do not use “Example” or “Examples” as a normal section heading (e.g. in module level docs): only use it as a NumPy doc Section. It will not be rendered properly, and will mess up sectioning.
When writing multiple common names in one line, Sphinx sometimes tries to reference the first name. In that case, you have to split the names across multiple lines. See below for an example:
Parameters ---------- n_atoms, n_residues : int numbers of atoms/residues
We are using MathJax with sphinx so you can write LaTeX code in math tags.
In blocks, the code below
#<SPACE if there is text above equation> .. math:: e^{i\pi} = -1
renders like so:
\[e^{i\pi} = -1\]Math directives can also be used inline.
We make use of the identity :math:`e^{i\pi} = -1` to show...
Note that you should always make doc strings with math code raw python strings by prefixing them with the letter “r”, or else you will get problems with backslashes in unexpected places.
def rotate(self, R): r"""Apply a rotation matrix *R* to the selection's coordinates. :math:`\mathsf{R}` is a 3x3 orthogonal matrix that transforms a vector :math:`\mathbf{x} \rightarrow \mathbf{x}'`: .. math:: \mathbf{x}' = \mathsf{R}\mathbf{x} """
See also
See Stackoverflow: Mathjax expression in sphinx python not rendering correctly for further discussion.
Documenting changes
We use reST constructs to annotate additions, changes, and deprecations to the code so that users can quickly learn from the documentation in which version of MDAnalysis the feature is available.
A newly added module/class/method/attribute/function gets a versionadded directive entry in its primary doc section, as below.
.. versionadded:: X.Y.Z
For parameters and attributes, we typically mention the new entity in a versionchanged section of the function or class (although a versionadded would also be acceptable).
Changes are indicated with a versionchanged directive
.. versionchanged:: X.Y.Z
Description of the change. Can contain multiple descriptions.
Don't assume that you get nice line breaks or formatting, write your text in
full sentences that can be read as a paragraph.
Deprecations (features that are not any longer recommended for use and that will be removed in future releases) are indicated by the deprecated directive:
.. deprecated:: X.Y.Z
Describe (1) alternatives (what should users rather use) and
(2) in which future release the feature will be removed.
When a feature is removed, we remove the deprecation notice and add a versionchanged to the docs of the enclosing scope. For example, when a parameter of a function is removed, we update the docs of the function. Function/class removal are indicated in the module docs. When we remove a whole module, we typically indicate it in the top-level reST docs that contain the TOC tree that originally included the module.
Writing docs for abstract base classes
MDAnalysis contains a number of abstract base classes, such as AnalysisBase. Developers who define new base classes, or modify existing ones, should follow these rules:
The class docstring needs to contain a list of methods that can be overwritten by inheritance from the base class. Distinguish and document methods as required or optional.
The class docstring should contain a minimal example for how to derive this class. This demonstrates best practices, documents ideas and intentions behind the specific choices in the API, helps to promote a unified code base, and is useful for developers as a concise summary of the API.
A more detailed description of methods should come in the method docstring, with a note specifying if the method is required or optional to overwrite.
See the documentation of MDAnalysis.analysis.base.AnalysisBase for an example of this documentation.
Adding your documentation to MDAnalysis
As with any contribution to an MDAnalysis repository, commit and push your documentation contributions to GitHub. If any fixes in the restructured text are needed, put them in their own commit (and do not include any generated files under docs/html). Try to keep all reST fixes in the one commit. git add FILE and git commit --amend is your friend when piling more and more small reST fixes onto a single “fixed reST” commit.
We recommend building the docs locally first to preview your changes. Then, create a pull request. All the tests in the MDAnalysis test suite will run, but only one checks that the documents compile correctly.
Viewing the documentation interactively
In the Python interpreter one can simply say:
import MDAnalysis help(MDAnalysis) help(MDAnalysis.Universe)
In ipython one can use the question mark operator:
In [1]: MDAnalysis.Universe?